Picture this: a Phase III trial has just wrapped up. Your regulatory affairs team is staring down an FDA submission deadline in six weeks. Somewhere across 47 clinical sites, thousands of Case Report Forms need to be verified, reconciled, and packaged. The adverse event log has 300+ entries, each requiring cross-referencing against the trial protocol. And the eCTD submission package? That's a 50,000-page document structure that needs to be audit-ready before it goes anywhere near the FDA's electronic gateway.
This is the document problem that doesn't get talked about enough in pharma. Not the science. Not the trials themselves. The paper mountain that sits between a completed trial and a drug approval.
Clinical trial document processing is one of the most high-stakes, error-prone, and labor-intensive workflows in any industry. Mistakes don't just cost money. They delay life-saving treatments, trigger FDA queries that push approval timelines out by months, and expose organizations to serious compliance risk. And yet, most pharma teams are still handling it with a combination of manual data entry, legacy OCR tools, and armies of data managers doing spot-checks by hand.
There's a better way to work. But first, it helps to understand exactly where the current approach breaks down.
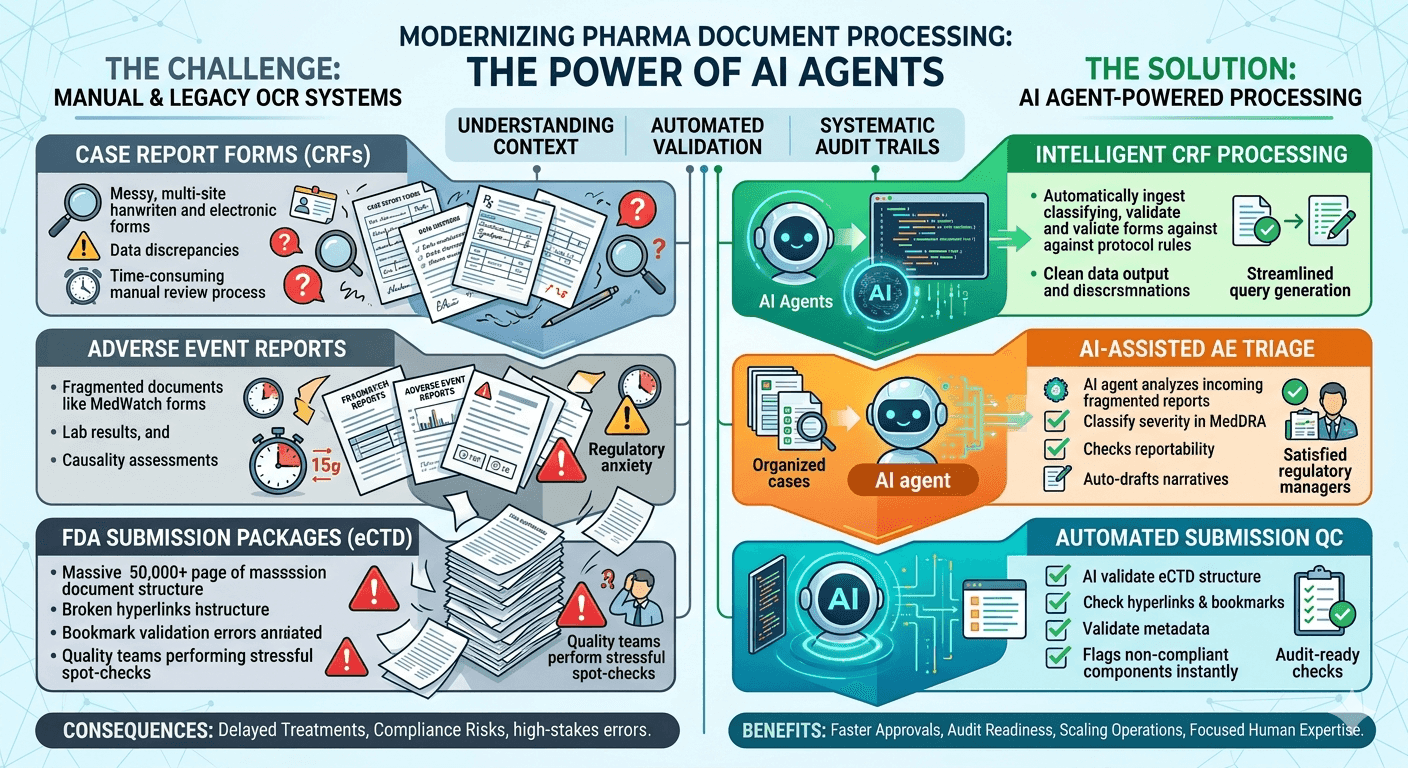
Three Document Types, Three Different Nightmares
The pharma regulatory document stack isn't monolithic. It's really three distinct workflows that each carry their own complexity, and each one demands precision that traditional document tools simply can't deliver.
Case Report Forms are the backbone of any clinical trial. Every patient visit, every measurement, every observation gets recorded in a CRF. In large multi-site trials, you're dealing with thousands of these documents coming in from dozens of locations, often in different formats, sometimes handwritten, sometimes partially completed in ePRO systems, and occasionally inconsistent with the source data in the trial database. The data manager's job is to catch every discrepancy before it touches the final dataset. That job has historically been done field-by-field, row-by-row, by human beings.
Adverse event reports carry even higher stakes. Under 21 CFR Part 312, sponsors are required to report serious and unexpected adverse events to the FDA within 15 calendar days. That clock starts from the moment the sponsor becomes aware of the event. The documentation trail for a single adverse event can involve the initial MedWatch form, a narrative summary, supporting lab results, a causality assessment from the investigator, and follow-up reports if the event evolves. Every piece of that needs to be captured, classified, and routed correctly. Miss a field or miscategorize severity, and you're looking at a reportability error with regulatory consequences.
FDA submission packages sit at the top of the complexity pyramid. An eCTD submission for a new drug application can contain hundreds of thousands of pages organized into a strict module structure. The clinical study reports alone can run 10,000 pages. Hyperlinks need to be checked. Bookmarks need to be validated. Documents need to be cross-referenced against the submission index. Quality teams spend weeks on pre-submission checks that still routinely miss errors the FDA's reviewer finds on the first pass.
These three workflows share one fundamental problem: the documents are complex, the data is structured in inconsistent ways, and the consequences of errors are severe. OCR alone doesn't solve this. It reads characters. It doesn't understand a CRF field that says "AE ongoing" means the adverse event report isn't complete. It doesn't know that a discrepancy between the CRF date and the source document date matters differently depending on whether it's a primary endpoint measurement.
Understanding the data in context is what makes pharma document processing genuinely hard.
Why Legacy OCR Tools Keep Failing Pharma Teams
The pharma industry spent years and significant money adopting OCR-based document management systems. The pitch was simple: scan your paper CRFs, extract the data, load it into your EDC system. Clean, automated, done.
The reality turned out to be considerably messier.
OCR tools work by recognizing character patterns. They're built for clean, standard text. Clinical trial documents are anything but. CRFs from different sites use different templates. Handwritten annotations appear in unexpected places. Checkboxes don't scan consistently. Tables shift across page layouts. Dates get formatted differently depending on which country the site is in. And critically, OCR tools have no ability to validate what they extract against the rules of the protocol or the logic of the underlying data model.
The result is extraction that's 80-90% accurate. That sounds reasonable until you do the math on a 10,000-page dataset. At 90% accuracy, you're generating hundreds of errors that need to be found and corrected manually. You've swapped one manual process for another, and the second one is harder because now someone has to find errors in extracted data rather than in original documents.
Adverse event processing is worse still. A single MedWatch form contains structured fields, free-text narrative sections, coded terminology that needs to map to MedDRA classifications, and causality assessments that require clinical judgment to interpret. Legacy systems can extract the structured fields with reasonable accuracy. The free text? That gets dumped into a notes field and handed back to a human.
This gap, between what document management systems claim to automate and what they actually automate, is where pharma teams lose enormous amounts of time and where compliance risk quietly builds up.
AI Agents Change the Processing Model
What changes with AI agents isn't just accuracy on extraction. It's the ability to understand documents in context and take actions based on that understanding.
An AI agent processing a CRF doesn't just read the fields. It understands what each field means in relation to the others. It can identify that a field marked "not applicable" contradicts a protocol requirement for that patient population. It can flag a visit date that's outside the protocol-specified window and route the discrepancy to the right data manager with the relevant protocol section attached. It can cross-reference the CRF against the source document and generate a query automatically when the values don't match.
For adverse event reports, the workflow shifts from manual classification to AI-assisted triage. The agent reads the incoming report, including the free-text narrative, classifies severity using MededRA terminology, checks the protocol's definition of expected events for that compound, and makes a preliminary determination about reportability. A regulatory affairs team member reviews and confirms, but they're now working with a structured summary and a recommended action rather than reading raw documents and making those determinations from scratch.
FDA submission packages benefit from a different kind of intelligence. The agent can parse the eCTD structure, validate hyperlinks and cross-references, check document metadata against submission requirements, and flag missing or non-compliant components before the package ever gets to pre-submission review. Teams that used to spend three weeks on manual QC checks are completing that work in days, and catching more issues in the process.
The shift isn't about removing humans from the loop. It's about changing where humans spend their time. Instead of reading and classifying, they're reviewing and deciding. The judgment-heavy work that actually requires their expertise stays with them. The pattern recognition and rules-based validation that a machine can do more accurately and consistently than any human gets automated.
What This Looks Like for CRF Processing Specifically
CRF processing is a good place to see how dramatically the workflow changes in practice.
In a traditional setup, CRFs come in from sites, either as paper scans or as exports from EDC systems. Data managers work through them manually, checking for completeness, coding terminology, and protocol deviations. Queries get raised in the EDC system. Sites respond. Managers review the responses. This cycle can run for months in a large trial, often extending well past database lock timelines.
With AI-powered processing, CRFs are ingested as they arrive. The system classifies each form by type and visit, extracts all structured fields, and immediately runs a battery of validation checks against the protocol rules loaded in the system. Protocol deviations, missing mandatory fields, out-of-range values, and date inconsistencies all get flagged automatically and documented with specific references to the relevant protocol sections.
Query generation becomes semi-automated. The agent drafts the query text, references the relevant CRF fields and protocol sections, and assigns severity. The data manager reviews the queue and approves or modifies queries rather than writing them from scratch. Resolution tracking happens in the same system, so there's a complete audit trail from initial discrepancy to final resolution.
Database lock timelines compress. What used to take 90 to 120 days of data cleaning post-database transfer often comes down to 30 to 45 days because discrepancies are caught and resolved in real time rather than in a post-trial cleanup phase.
The Adverse Event Reporting Timeline Problem
The 15-day reporting clock for serious adverse events is one of the most unforgiving regulatory requirements in pharma. It leaves almost no room for process inefficiency, and the consequences of missing it are serious.
The challenge is that the information required for a complete expedited report often comes in fragments. The initial report from the investigator may be incomplete. Lab values might arrive separately. The causality assessment might take days to obtain. Someone has to track all of this, chase down missing information, and assemble the complete package before the clock runs out, all while the regular workload of the safety department continues.
AI agents bring structure to this fragmented process. When an adverse event comes in, the agent creates a case record immediately, timestamps the awareness date, and starts tracking completeness against the MedWatch form requirements. As additional information arrives (by email, fax, or EDC), the system matches it to the open case and updates the completeness status. Automated reminders go out to investigators when information is still missing at day 7 or day 10. The narrative summary gets drafted by the AI based on the available data, ready for medical review.
The regulatory affairs team still owns every decision. But they're working from a structured, pre-assembled case record rather than building it manually from scattered inputs. The 15-day deadline becomes manageable rather than a regular source of regulatory anxiety.
Audit Readiness as an Output, Not a Project
One of the quieter benefits of AI-powered pharma document processing is what it does to audit preparation.
In most organizations, regulatory audits trigger a separate project. Someone has to pull together documentation, verify completeness, check that all queries are resolved, and make sure the audit trail is intact. This takes weeks of work from regulatory affairs staff who should be focused on other things, and it creates anxiety because the preparation itself can uncover gaps that then need to be addressed.
When document processing is AI-driven and systematic, audit readiness becomes a continuous state rather than a periodic project. Every document is processed the same way, every time. The validation rules are consistent. The audit trail is generated automatically. Query resolution is tracked in the system. When an auditor asks for the documentation behind a specific adverse event or a specific CRF entry, it's retrievable in minutes, not hours.
FDA inspectors have increasingly made it clear that they're looking at systems, not just outcomes. A sponsor that can demonstrate consistent, validated document processing with clear audit trails is in a fundamentally different position from one that's relying on manual processes with retrospective documentation. The system becomes part of the compliance evidence.
From Submission Prep to Ongoing Regulatory Intelligence
For pharma organizations doing multiple submissions simultaneously or managing ongoing post-market reporting obligations, the document processing challenge doesn't end with approval. It becomes a permanent operational requirement.
Periodic Safety Update Reports, post-market surveillance documentation, protocol amendments, and annual IND progress reports all generate ongoing document processing work. The same AI-powered workflow that handles Phase III CRFs can handle post-market case processing. The same submission package validation that works for an NDA works for a supplemental application.
This is where Artificio's model differs from point solutions. Instead of deploying separate tools for CRF processing, adverse event management, and submission preparation, pharma teams get a unified platform that handles the full regulatory document lifecycle. The AI agents understand the relationships between documents across different parts of the workflow. A safety signal that shows up in adverse event reports can be cross-referenced against CRF data automatically, giving safety teams a more complete picture without requiring manual data pulls across multiple systems.
The volume of regulatory documentation in pharma isn't going to shrink. The FDA keeps adding requirements. International harmonization means submissions need to meet multiple regulatory standards simultaneously. Post-market obligations grow as products age. The only sustainable path is a processing infrastructure that can scale with that volume while maintaining the accuracy and consistency that regulators expect.
Pharma teams that build that infrastructure now aren't just solving today's backlog. They're building the operational foundation that makes the next drug development cycle faster, cleaner, and more defensible when regulators come knocking